專注國內外醫(yī)療器械咨詢服務
注冊認證 · 許可備案 · 體系輔導 · 企業(yè)培訓
400-888-7587
0755-86194173、13502837139、19146449057
020-82177679、13602603195
四川:028-68214295、15718027946
湖南:0731-22881823、15013751550

注冊認證 · 許可備案 · 體系輔導 · 企業(yè)培訓
400-888-7587
0755-86194173、13502837139、19146449057
020-82177679、13602603195
四川:028-68214295、15718027946
湖南:0731-22881823、15013751550
作為發(fā)達國家中的一員,韓國的醫(yī)療器械市場規(guī)模是不可小覷的,然而與美國等其他發(fā)達國家相比,目前韓國醫(yī)療器械產業(yè)的國際競爭力仍處于較低水平,相關數據顯示,其國內半數以上的獲準醫(yī)療器械都源于韓國境外的國家。那么本期文章我們就來跟大家分享一下韓國醫(yī)療器械注冊的相關知識。 韓國根據產品對人體的潛在危害:1)與人體接觸的持續(xù)時間;2)侵入性程度;3)是否向患者提供藥品或能量;4)是否對患者有生物學影響,將醫(yī)療器械分為Ⅰ、Ⅱ、Ⅲ、Ⅳ四個風險等級。
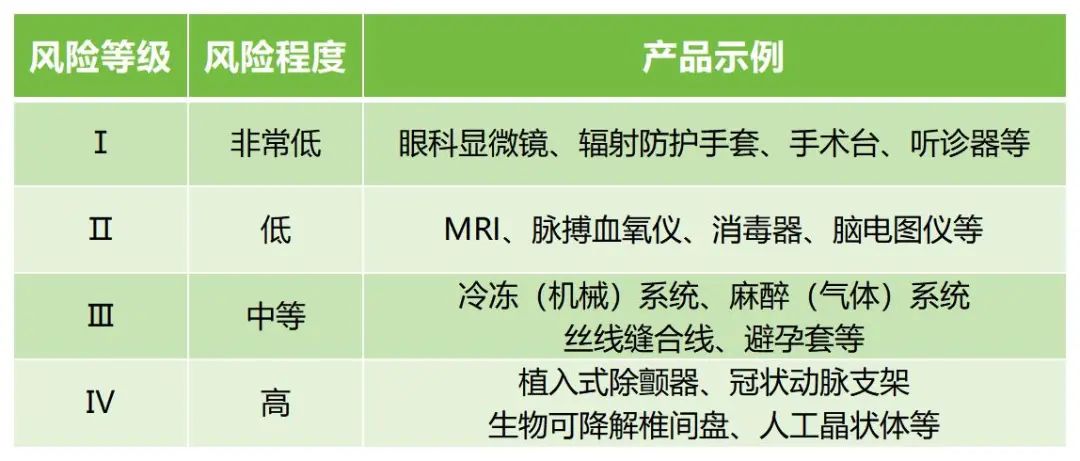
↑ 具體產品示例
1. 查閱Regulation on Medical Device Groups and Class by Group的Attachment,法規(guī)中規(guī)定了分類規(guī)則以及產品的代碼和風險等級,可以根據產品定義/用途等判斷產品的等級,我們隨附了示例圖,紅框的數字為產品的風險等級。 2. 在韓國已注冊產品的數據庫(可聯系我們獲取鏈接)中,用產品名稱(英文/韓文)進行檢索,查找類似產品在MFDS中的分類。 上述方法需一并查詢以確定產品在韓國的分類。當然,您也可以直接聯系我們幫您查詢~
在韓國,Ⅰ類產品一般只需在系統(tǒng)上做簡單的Notification登記即可,有SE Device(即實質等同產品)的Ⅱ類產品,需遞交技術資料進行NIDS認證;而對于NSE Device(即沒有實質等同產品)的Ⅱ類產品以及Ⅲ、Ⅳ類產品,則需要由MFDS批準。不同類別產品所需提交的注冊資料不同,具體可聯系我們咨詢。
和大部分國家或地區(qū)一樣,在韓國沒有實體辦公場所的公司必須任命一名韓國許可持有人(Korean License Holder)來協(xié)調他們向MFDS注冊醫(yī)療器械的事務。KLH控制醫(yī)療器械注冊并幫助韓國境外制造商遵守韓國良好生產規(guī)范(KGMP)要求,并且其名字會出現在制造商的MFDS醫(yī)療器械注冊證書(產品審批證書)上。產品獲批后,KLH需負責進口醫(yī)療器械的年度報告事宜,并且接受MFDS的突擊審核。
一般情況下,企業(yè)會選擇在韓國的分銷商作為證書持證人,但選擇沒有銷售合作并有能力協(xié)助產品注冊的韓國企業(yè)作為持證人,在后續(xù)需要更換經銷商時可能會更加便捷。
在韓國,Ⅱ、Ⅲ、Ⅳ類醫(yī)療器械的制造商都需要符合KGMP(Korean Good Manufacturing Practice)的要求,KGMP要求與ISO 13485類似。
KGMP證書是頒發(fā)給進口商(Importor)而不是制造商(Manufacturer), 證書每3年需更新一次,企業(yè)要在在證書過期前的90天更新。
通常對于高風險的Ⅲ、Ⅳ類醫(yī)療器械,由監(jiān)管當局MFDS審核制造商的質量管理體系,而Ⅱ類和部分Ⅲ、Ⅳ類的醫(yī)療器械交由第三方機構審核。
審核現場的語言要求是韓語,因此企業(yè)需要提前準備韓語翻譯。審核周期大約在9-12個月。
![]()
醫(yī)療器械注冊咨詢認準金飛鷹 深圳:0755-86194173 廣州:020 - 82177679 湖南:0731-22881823 四川:028 - 68214295
全國服務熱線:400-888-7587
總部地址:深圳市南山區(qū)前海路3101號-90振業(yè)國際商務中心2401
廣州地址:廣州市黃埔開發(fā)區(qū)科學大道50號綠地中央廣場A3棟1906
四川地址:成都市高新區(qū)益州大道中段722號2棟1單元12層1209
湖南地址:株洲市天元區(qū)泰山路238號東帆國際大廈1823-1824
湖北地址:武漢市東湖新技術開發(fā)區(qū)關東街道高新二路9號北辰光谷里8A棟1607
江蘇地址:蘇州市工業(yè)園區(qū)唯華路5號君風生活廣場17棟918
廣西地址:玉林市玉州區(qū)仁厚路與緯二路交叉口玉林中醫(yī)藥健康產業(yè)園1號樓516
海南地址:海口市國家高新技術產業(yè)開發(fā)區(qū)南海大道266號創(chuàng)業(yè)孵化中心A樓5層A26
重慶地址:重慶兩江新區(qū)水土新城生命科技城展示中心2樓
版權所有:深圳市金飛鷹企業(yè)管理顧問有限公司
備案號:粵ICP備16108171號 技術支持:深度網


 返回
返回